Can a 70 Year Old Man Have Cystic Fibrosis
Overview
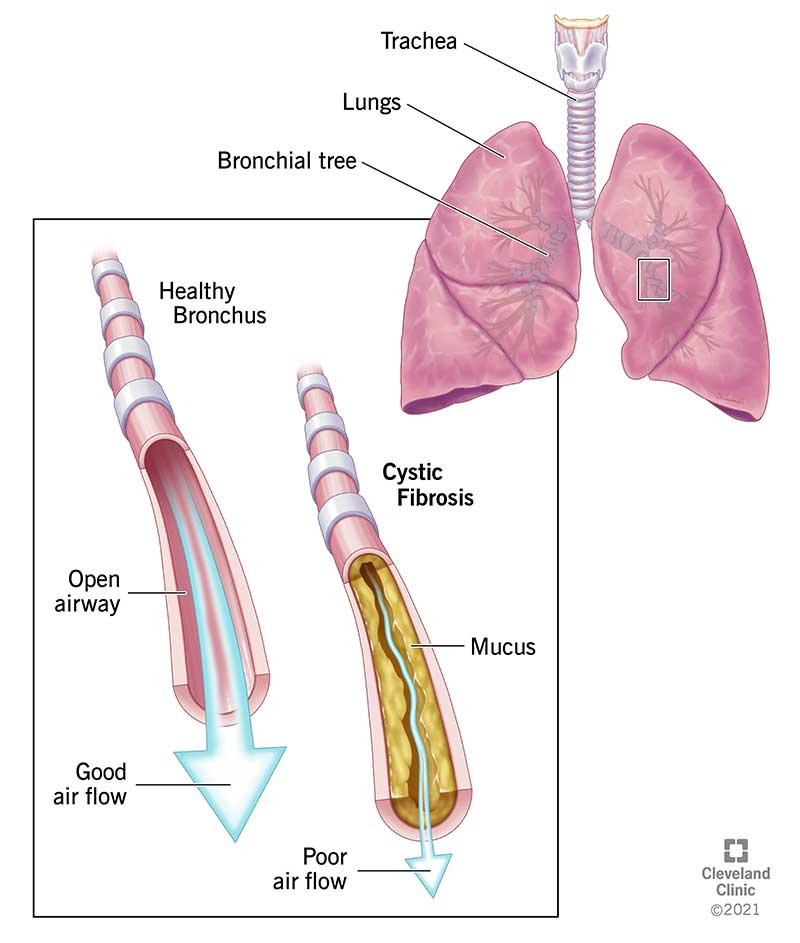
What is cystic fibrosis (CF)?
Cystic fibrosis (CF) is a genetic (inherited) disease that causes sticky, thick mucus to build up in organs, including your lungs and pancreas.
If yous don't take CF, the mucus that lines organs and body cavities, such as your lungs and nose, is slippery and watery. If yous exercise have CF, thick mucus clogs the airways and makes it difficult to breathe.
Mucus also blocks the ducts in the pancreas, causing problems with digesting nutrient. Babies and children who accept CF might not be able to absorb enough nutrients from nutrient. CF, which is chronic (long-lasting) and progressive (getting worse over time), also affects your liver, sinus, intestines and sex activity organs.
There's also a form of disease called "atypical cystic fibrosis." It's different from classic CF because information technology's a milder course and may but impact i organ. The other "singular" thing well-nigh it is that it unremarkably comes on much later in life. "Typical" or archetype CF generally shows up in the first few years of a child's life.
How common is cystic fibrosis (CF)?
Amongst white children in the U.S., the rate of CF cases is 1 in 2,500 to iii,500 newborns. CF affects nigh 1 in 17,000 Black newborns and ane in 31,000 newborns of Asian descent.
Symptoms and Causes
What causes cystic fibrosis (CF)?
Cystic fibrosis is genetic. People who accept CF inherit two faulty genes, one from each parent. CF is said to be recessive because you demand to take two gene variants to take the condition itself. (An older name for gene variant is gene mutation.)
Your parents don't have to have cystic fibrosis for yous to take CF. In fact, many families don't have a family unit history of CF. If your family doesn't have a history of cystic fibrosis, the person with the gene variant is called the carrier. About 1 in 31 people in the U.South. is a carrier who is free of CF symptoms.
What are the symptoms of cystic fibrosis (CF)?
Classic cystic fibrosis
Children who have archetype CF have the following symptoms:
- Failure to thrive (disability to proceeds weight despite having a good appetite and taking in enough calories).
- Loose or oily stools.
- Trouble animate.
- Recurrent wheezing.
- Frequent lung infections (recurrent pneumonia or bronchitis).
- Recurrent sinus infections.
- A nagging cough.
- Deadening growth.
Atypical cystic fibrosis
People with atypical cystic fibrosis may exist adults past the time they're diagnosed with atypical CF. Respiratory signs and symptoms may include:
- Chronic sinusitis.
- Breathing bug, maybe diagnosed as asthma or chronic obstructive pulmonary affliction (COPD).
- Nasal polyps.
- Frequent bouts of pneumonia.
Other signs and symptoms of singular CF may include:
- Dehydration or heat stroke that reveals aberrant electrolyte levels.
- Fertility problems.
- Diarrhea.
- Pancreatitis.
- Unintended weight loss.
Diagnosis and Tests
How is cystic fibrosis (CF) diagnosed?
In most cases, CF is diagnosed during babyhood. Doctors diagnose CF with a thorough evaluation and past using different tests. These include:
- Newborn screening: Your healthcare provider takes a few drops of blood from a heel prick, usually while your newborn is in the hospital, and places the drops on a special card called a Guthrie card. The screening looks for a list of conditions, including CF. Every U.S. state requires the testing of newborns at nativity and a few weeks later.
- Sweat test : The sweat test measures the amount of chloride in the body's sweat, which is college in people who take CF. In the test, your healthcare provider spreads a chemical called pilocarpine on your skin, then applies a minor corporeality of electric stimulation to encourage the sweat glands to produce sweat. Your provider so collects the sweat in a plastic coil or on a piece of filter paper or gauze. People of any age can accept a sweat examination. It's not painful and does not apply a needle. This is the most conclusive examination for CF.
- Genetic tests: Claret samples are tested for the genes that cause CF.
- Chest X-rays : Your healthcare provider will social club X-rays of the chest are used to support or confirm CF, just a chest X-ray isn't the only test needed to confirm a diagnosis. Other tests must exist done.
- Sinus X-rays: Equally with chest X-rays, sinus X-rays can confirm CF in people who show sure symptoms. Other forms of testing are used along with sinus X-rays.
- Lung function tests: The most mutual lung function test uses a device chosen a spirometer. You breathe in completely, and so push the inhaled breath into the mouthpiece of the spirometer.
- Sputum culture: Your healthcare provider takes a sample of your sputum (spit) and tests it for leaner. Certain bacteria, such as Pseudomonas, are most normally plant in people who have CF.
- Nasal potential difference (NPD): This test uses a voltmeter and electrodes placed in two places in your nose and i identify exterior of your olfactory organ to measure the electricity generated by the transfer of ions in solution across the nasal tissue. The exam uses three different types of solutions.
- Abdominal current measurement (ICM): Yous'll have to have a biopsy of rectal tissue for this test. The tissue is made to secrete chloride, which is then measured.
In people who take singular cystic fibrosis, the sweat examination may exist normal in terms of the levels of chloride. Some people with atypical CF may have been born before testing became routine. Your provider may social club NPD and ICM tests when the diagnosis is questionable.
Management and Treatment
How is cystic fibrosis (CF) treated?
You'll probably take a healthcare team that includes a specialist in cystic fibrosis and many other types of caregivers. There is no cure for cystic fibrosis, but your team will help you manage the disease. The major focus of management is keeping your airways clear. Your provider will also prescribe medicine when needed.
Keeping airways clear
You tin can assist to proceed your airways articulate if you have cystic fibrosis in a number of ways:
- Yous can larn special ways of coughing and breathing.
- You lot can use devices that fit into your mouth or therapy vests that rely on vibrations to loosen mucus.
- You can learn something chosen breast physical therapy, as well known as postural drainage and percussion to loosen mucus. With this method, you movement into certain positions so that your lungs tin bleed. Another person claps their hand on your chest and/or your dorsum to help loosen the mucus. You might combine this with coughing.
Medications for cystic fibrosis
Your provider may prescribe these medicines, which won't cure CF, but which will help you lot in sure situations. They include:
- Antibiotics to treat lung infections or prevent them.
- Inhaled bronchodilators to brand breathing easier by opening and relaxing your airways.
- Inhaled medicine to make fungus thinner and easier to get rid of.
- Anti-inflammatory drugs, including steroids and non-steroidal anti-inflammatories.
- Medications to treat the crusade of cystic fibrosis in people with certain gene variants.
- Pancreatic enzymes to assistance in digestion.
- Stool softeners to assistance with constipation.
Surgeries for cystic fibrosis
Yous may need surgery for cystic fibrosis or 1 of its complications. These might include:
- Surgery on your nose or sinuses.
- Bowel surgery to remove blockages.
- Transplant surgery, including a double lung transplant or a liver transplant.
Why is a high-calorie, high-fat diet needed for people with cystic fibrosis (CF)?
People with cystic fibrosis have nutritional needs that aren't the same as the needs of people without CF. People with CF may need 1.5 to 2 times the number of calories every bit people without CF. You need the extra calories if yous accept CF because you use more energy than other people to breathe, fight lung infections and maintain your strength.
You besides demand more calories and fat because cystic fibrosis stops the digestive enzymes fabricated past your pancreas from working completely. This means nutrients and fats from foods aren't fully captivated past your intestines.
Although the enzyme capsules that are taken before all meals and snacks helps digest fats, proteins and starches, a sure corporeality of nutrients and fats don't get absorbed. If your body doesn't absorb enough fats, so fatty-soluble vitamins aren't being fully absorbed either, and these vitamins are needed to protect the lungs.
It's too important to stress that people with cystic fibrosis should keep a higher than normal weight starting in early babyhood. Researchers take shown that young people with CF who maintain a higher weight grow faster and taller upwardly to puberty and once more grow taller when they hitting their growth spurt at puberty.
Immature people with CF who started life at a lower weight did not grow equally many inches, started puberty at a later age and never got that same puberty growth spurt. Reaching your full genetic potential — getting as tall as possible with lungs as large as possible — is another reason why higher-than-normal weight in immature people with CF is and so important.
Another common misbelief is that salt (sodium) is unhealthy for all people. This isn't true for children and adults with CF. People with CF lose a lot of salt in their sweat. Although there's not a ready standard, healthcare providers generally tell people with CF to swallow salty foods. This is truthful particularly during hot, humid weather and practise. If you have CF, y'all can probably add salt to meals and snacks as desired. Enquire your provider or a registered dietician well-nigh the corporeality of salt you need each twenty-four hour period.
What are the complications of cystic fibrosis (CF)?
The complications of CF include the following:
- Adults who have CF tin can have problems with animate, digestion and their reproductive organs.
- The thick mucus present in people who have CF tin hold bacteria, which tin lead to more infections.
- People who have CF have a higher run a risk of developing diabetes or the os-thinning conditions like osteopenia and osteoporosis.
- Men who accept CF are non able to father children without the aid of alternative reproductive applied science. Women who have CF can have a decrease in fertility (the power to have children) and complications in pregnancy.
Prevention
How can I forbid cystic fibrosis?
Y'all can't prevent cystic fibrosis considering it's an inherited condition. If you or your partner have any kind of family history, you may desire to speak to a genetic counselor before yous decide to accept children.
Outlook / Prognosis
What is the prognosis (outlook) for people who accept cystic fibrosis (CF)?
There is no cure for CF and it cannot exist prevented. Yet, new handling methods help children who take CF live well into adulthood and have a meliorate quality of life.
Therapies are nearly helpful when CF is diagnosed early on, which is why newborn screening is then important. These therapies include treating infections, trying to prevent weight loss and seeing a CF specialist frequently. The addition of cystic fibrosis transmembrane conductor regulator (CFTR) modulator therapy at a young age seems to be very benign and may improve long-term health.
According to information from the Cystic Fibrosis Foundation Patient Registry, more than than half of people built-in with CF betwixt 2022 and 2022 are expected to live to age 46 or longer.
People with atypical cystic fibrosis tend to have longer life expectancies than those with classic CF.
Living With
How do I take care of myself if I have cystic fibrosis?
An adult with cystic fibrosis has different needs than a child with CF. If yous're a parent of a child with CF or if you lot're an adult with CF, you tin do a lot to promote a salubrious life. This includes developing and following recommendations from a treatment plan developed with your healthcare squad.
Follow suggestions from your providers about eating enough, eating well and exercising wisely. Ask your provider if pulmonary rehabilitation would be a good idea for you.
Take care to forbid infections past distancing yourself from people who are sick. Practice expert hand washing techniques. Get the vaccines that your providers say are needed.
Follow any recommended schedule of appointments with your provider and other members of your healthcare team. If you need assistance with social or emotional issues, reach out to your squad and examine your options.
Decide if you lot'd like to be function of a clinical trial. Enquire your provider to betoken you in the right direction to be a participant.
Frequently Asked Questions
Is cystic fibrosis contagious?
No. Cystic fibrosis isn't contagious. It's a genetic disorder, not an infection. You can't catch it from anyone and you lot tin't give information technology to anyone. If you accept it, though, you need to be careful if y'all're exposed to infections.
Can you become cystic fibrosis at any age?
Most cases of cystic fibrosis are plant during the outset few years of life. However, it's possible to get an adult then exist diagnosed every bit having CF.
Is cystic fibrosis a concluding illness?
In the past, cystic fibrosis was considered to be a fatal illness. People who had it died in babyhood. This is no longer true. Today, almost children who accept CF grow upwards to be adults with CF.
A note from Cleveland Dispensary
If yous have cystic fibrosis or your child has CF, y'all know that this genetic disorder requires lifelong direction. As with many illnesses, beingness diagnosed early and getting treatment early unremarkably results in the best outcomes. Work with your healthcare squad, or your child'southward healthcare squad, to discover ways to stay salubrious. You lot may accept access to more resources than you realize. Inquiry is ongoing and scientists are working toward fifty-fifty better outcomes.
Source: https://my.clevelandclinic.org/health/diseases/9358-cystic-fibrosis
0 Response to "Can a 70 Year Old Man Have Cystic Fibrosis"
Post a Comment